") 聚類分析中的機器學習與統(tǒng)計方法綜述(一)
聚類分析中的機器學習與統(tǒng)計方法綜述(一)
01
概況
單細胞轉(zhuǎn)錄組測序(scRNA-seq)技術(shù)能夠?qū)毎褐械拿恳粋€細胞進行大規(guī)模的全轉(zhuǎn)錄組分析。它的核心分析是將單細胞聚類,以揭示細胞亞型,并根據(jù)細胞之間的關(guān)系推斷細胞譜系。本文綜述了在過去幾年間發(fā)展起來的,用于單細胞轉(zhuǎn)錄組分析中聚類的機器學習和統(tǒng)計方法,重點介紹了如何將一些常見的聚類方法,如層次聚類、基于圖的聚類、混合模型、k-means、集成學習、神經(jīng)網(wǎng)絡(luò)和基于密度的聚類等加以調(diào)整及應(yīng)用,從而解決單細胞轉(zhuǎn)錄組數(shù)據(jù)分析中的獨特挑戰(zhàn),例如低表達基因的缺失,轉(zhuǎn)錄本的不均勻覆蓋,以及由技術(shù)偏差和不相關(guān)的混雜生物變異所帶來的細胞標記的失真。我們評價了標準化、dropouts推測以及降維等預處理步驟如何提高聚類效果。此外,我們還將介紹一些能夠?qū)r間序列樣本和多個細胞群進行聚類并且檢測罕見細胞類型的新方法。最后,本文對部分開發(fā)用于單細胞轉(zhuǎn)錄組聚類分析的軟件進行了實驗和比較,以評估其性能和效率,為未來的數(shù)據(jù)分析提供一定的指導和方向。
02
介紹
細胞的轉(zhuǎn)錄組分析可以捕捉基因的表達活性,從而揭示細胞的身份和功能。在傳統(tǒng)的bulk-RNA測序中,轉(zhuǎn)錄組是通過從生物樣本中收集的大量細胞轉(zhuǎn)錄水平的平均值來測量的,這些平均后的表達值被用于基因共表達模塊的識別和樣本聚類。由于忽略了單個細胞的特性,這些傳統(tǒng)的方法無法在單細胞分辨率上研究重要的生物學問題,如細胞在早期發(fā)育過程中的不同功能角色、復雜組織中的不同細胞類型和細胞譜系關(guān)系。目前,scRNA-seq技術(shù)已廣泛用于量化單個細胞中的mRNA水平。在單細胞轉(zhuǎn)錄組的實驗操作中,使用不同的捕獲方法(如FACS,F(xiàn)luidigm C1,microdroplet microfluidics)分離單細胞,然后對RNA進行逆轉(zhuǎn)錄并擴增測序。單細胞轉(zhuǎn)錄組的應(yīng)用已經(jīng)帶來了重要的生物學見解和發(fā)現(xiàn),例如,對癌癥中腫瘤異質(zhì)性的理解。
細胞聚類是單細胞轉(zhuǎn)錄組數(shù)據(jù)分析中識別細胞亞群結(jié)構(gòu)的必要步驟,然而目前仍然存在一些挑戰(zhàn)。首先,由細胞的自身特征(如細胞所處周期階段、細胞大小)和技術(shù)(捕獲方法、捕獲效率、PCR擴增、測序深度等)引入的技術(shù)噪音和偏差。這些噪音和偏差將導致轉(zhuǎn)錄組的基因覆蓋極度不均勻,從而造成零覆蓋區(qū)域和dropouts的產(chǎn)生。另外,當一個隊列的多個樣本同時進行分析時,樣本間的技術(shù)偏差和變異將會主導細胞的聚類,導致細胞群體的形成更偏向于不同樣本來源而非細胞類型,即批次效應(yīng)。
在本文中,我們回顧了最近發(fā)展的用于提升單細胞轉(zhuǎn)錄組聚類效果或其相關(guān)的統(tǒng)計和機器學習方法。這些方法涉及:(1)用于基因表達值的標準化、dropouts推測、數(shù)據(jù)降維以及細胞特異Marker鑒定的數(shù)據(jù)預處理方法;(2)傳統(tǒng)的聚類算法,包括基于劃分的聚類、層次聚類、混合模型、基于圖的聚類、基于密度的聚類、神經(jīng)網(wǎng)絡(luò)、集成聚類和近鄰傳播聚類等;(3)在時間序列樣本和多個批次的細胞群中進行聚類并檢測罕見細胞類型的新方法。我們還討論了單細胞轉(zhuǎn)錄組聚類分析中的幾個重要方面,包括細胞間相似性度量,特征值提取和單細胞聚類結(jié)果的評估。此外,我們對十多個軟件包進行了比較,以評估它們在大規(guī)模單細胞轉(zhuǎn)錄組數(shù)據(jù)集上的聚類性能和效率。最后,我們對聚類分析中存在的一些挑戰(zhàn)進行了討論。
03
數(shù)據(jù)的預處理
在單細胞轉(zhuǎn)錄組數(shù)據(jù)的聚類分析中,數(shù)據(jù)預處理對于減少技術(shù)變異和噪聲(如捕獲效率低、擴增偏差、GC含量、總RNA含量和測序深度的差異等)以及建庫和測序過程中產(chǎn)生的dropouts至關(guān)重要。高維的基因表達矩陣通常需要經(jīng)過標準化及降維映射到低維空間中,一些計算方法還利用到統(tǒng)計學和數(shù)學方法來解決dropouts事件。
標準化
原始的單細胞轉(zhuǎn)錄組數(shù)據(jù)通常從兩個層面進行標準化:細胞的標準化和基因的標準化。細胞的標準化是為了消除擴增偏差和其他細胞特異性的效應(yīng),可以通過常用的reads計數(shù)標準化方法實現(xiàn),如FPKM、RPKM、TPM等。基于UMI建庫的實驗方案,理論上已經(jīng)避免了與擴增或測序深度相關(guān)的誤差,因為被相同UMI標記的reads只會統(tǒng)計一次。然而,由于測序文庫通常是不飽和的,標準化對于該類型的數(shù)據(jù)也是有效的。細胞標準化的另一個方法是使用“spike-in”,它的基本思想是,由技術(shù)原因帶來的誤差對于內(nèi)外源基因的影響是相同的。另外,使用對數(shù)轉(zhuǎn)換進行原始計數(shù)值的處理也非常常見。
基因標準化的目的是為了防止一些高表達基因主導了分析。常用的基因標準化方法如,在PCA中包含的z-score標準化。從過往的經(jīng)驗中可以看到,基因的標準化可以提高算法的收斂和聚類效果。值得注意的是,數(shù)據(jù)的標準化處理將會使其失去原本基因表達的相對尺度,并且由于表達值的平移,造成表達矩陣變得不那么稀疏,這可能會影響到大規(guī)模數(shù)據(jù)集的聚類結(jié)果。
在SINCERA包中,對基因的標準化方法即是z-score,對細胞的標準化則是使用截尾均值(Trimmed mean)。一些工具會執(zhí)行更為特殊的標準化。例如,BISCUIT通過學習代表技術(shù)誤差的參數(shù),在聚類過程中進行迭代標準化;RaceID將每個細胞內(nèi)的總表達計數(shù)標準化到所有細胞表達計數(shù)的中位值。
此外,如果基因或者細胞顯現(xiàn)出極低的表達信號(基因表達值過低或者細胞表達基因過少),通常會將其移除,因為它們往往代表著虛假信號。在不同的研究中,為去除低表達基因和細胞建立了不同的閾值,這主要根據(jù)分析中囊括的細胞和基因的數(shù)量而有所不同。例如,scVDMC對PBMC樣本的處理中,表達值低于3的基因和總表達計數(shù)值小于200的細胞都將被去除。
雖然基因和細胞的標準化在目前大多數(shù)的單細胞數(shù)據(jù)分析流程中是常見的,但關(guān)于其對聚類結(jié)果的影響仍存在一些爭論。一項研究的分析表明,基于bulk的標準化方法在單細胞上的應(yīng)用可能會對其分析產(chǎn)生嚴重的不良后果,例如在聚類前進行的高變基因的檢測。相同的,也有研究表明,通過中位數(shù)或者“spike-in”進行標準化無法解決dropouts存在的問題,反而可能消除每種細胞類型特有的生物隨機性,這兩者都會導致潛在的細胞類型的不恰當聚類或表征。
通過下面的例子,我們可以認識到標準化的重要性。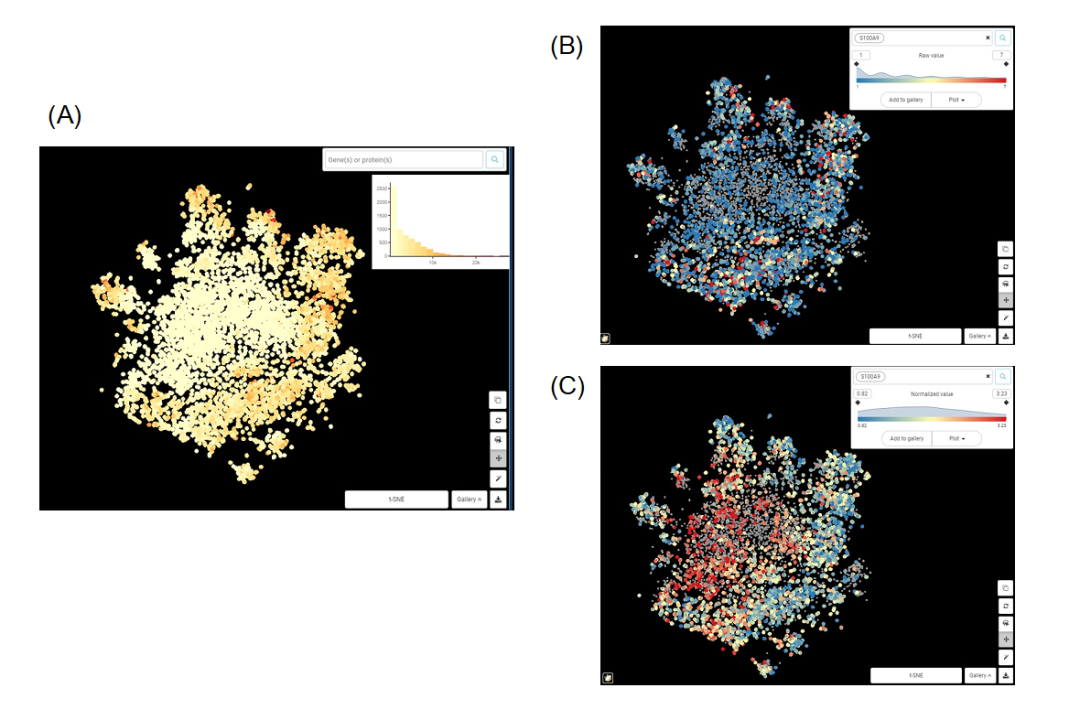
Figure 1. 巨噬細胞群t-SNE圖 來自Zilionis等人數(shù)據(jù)集的巨噬細胞群t-SNE圖。(A)依據(jù)總計數(shù)值上色;(B)依據(jù)基因S100A9原始計數(shù)值上色;(C)依據(jù)標準化后的S100A9的表達值上色。
從圖1A,B很容易看出,S100A9的原始表達值與總計數(shù)高度相關(guān),兩個圖的中心區(qū)域計數(shù)和表達量都較低,而外圍區(qū)域計數(shù)和表達量較高。我們能得出的唯一結(jié)論是,當細胞中捕獲的轉(zhuǎn)錄本總量增加時,S100A9轉(zhuǎn)錄本的數(shù)量也會增加。這顯然沒什么意義。而在圖1C中,經(jīng)過標準化后的S100A9表達值與總計數(shù)之間似乎沒有相關(guān)性。我們可以說,S100A9表達的差異不依賴于測序深度等技術(shù)噪音,而應(yīng)該來自(主要)生物因素。
Dropout
單細胞轉(zhuǎn)錄組數(shù)據(jù)中一個重要的技術(shù)誤差被稱為“dropouts”。Dropout事件是指在反轉(zhuǎn)錄過程中由于缺失或轉(zhuǎn)錄本表達過低而導致基因未表達的錯誤定量。先前的研究也表明,簡單的數(shù)據(jù)標準化并不能解決該問題。因此,一些聚類算法中包含了特定的機制以矯正dropouts。例如,Seurat通過跨細胞的基因共表達模式,在聚類前進行標記基因的挑選。
另外,也可以通過計算配對相似性來估算dropouts。CIDR便是在聚類前進行缺失值的填補。首先分析單細胞中可能出現(xiàn)的dropouts,識別每個細胞中的候選dropout基因,計算每個基因的dropout率;然后使用候選基因的dropout率來估算表達水平,即當dropout事件以高概率被識別時,檢測算法會從其它細胞的表達譜中對該基因的表達值進行填充;最后,利用矯正后的值計算細胞間的不相似度,進行層次聚類。Seurat和SNN-Cliq是基于共享最近鄰SNN來度量細胞相似性。已經(jīng)證明,在稀疏的高維數(shù)據(jù)中,SNN考慮到周圍的近鄰數(shù)據(jù)點,更適合應(yīng)用于存在dropouts的聚類分析。
在一個更復雜的概率圖模型中,BISCUIT明確估計了每個細胞中的基因表達,以及通過數(shù)據(jù)分布和先驗分布估算的代表技術(shù)和生物學變異的參數(shù)。其中,代表著未觀測到的基因真實表達水平的隨機變量被引入圖模型中并通過吉布斯抽樣來估算表達值。
降維
降維通常用于將高維基因表達數(shù)據(jù)投射到低維空間,使分析聚焦于低維空間中的相關(guān)信號,從而更好地實現(xiàn)數(shù)據(jù)的可視化、聚類分析等,幫助進行生物學解釋。當維數(shù)大于樣本數(shù)時,降維還有助于解決樣本不足的統(tǒng)計學問題。許多降維方法已經(jīng)應(yīng)用于單細胞轉(zhuǎn)錄組聚類算法,包括PCA、多維尺度變換(MDS)、t分布、隨機近鄰嵌入(t-SNE)、典型相關(guān)分析(CCA)、潛在狄利克雷分布(LDA)以及嵌入其他模型的降維等等。
PCA:將原本數(shù)據(jù)點映射到與協(xié)方差矩陣的最大特征值相關(guān)聯(lián)的特征向量(即主成分),以保留原始數(shù)據(jù)中的大部分方差。例如,pcaReduce在聚類前將表達矩陣映射到一個含有K-1個主成分的空間中;SC3使用PCA和拉普拉斯變換應(yīng)用于距離矩陣以獲得一致性矩陣并進行層次聚類。此外,在聚類之后,PCA也被廣泛應(yīng)用于二維或三維的數(shù)據(jù)可視化。PCA是一種基于假設(shè)數(shù)據(jù)為高斯分布的線性投影方法,為了捕捉數(shù)據(jù)中的非線性結(jié)構(gòu),可以使用核主成分分析與非線性核映射相結(jié)合。
MDS:也稱為主坐標分析(PCoA)。MDS將數(shù)據(jù)點映射到低維空間,通過最小化所有配對數(shù)據(jù)點的原始空間中的距離與投影空間中的距離之間的差值,從而在低維嵌入保持原始高維空間中的數(shù)據(jù)點之間的距離。CIDR便是使用MDS來計算細胞的不相似矩陣。MDS的優(yōu)點是在低維空間中保持原始的成對距離,易于實現(xiàn)非線性特征嵌入。然而,MDS不能擴展到大規(guī)模數(shù)據(jù),因為必須計算成對距離來最小化目標函數(shù)。
t-SNE:是一種將距離轉(zhuǎn)換為概率的方法。t-SNE構(gòu)造一個與原始空間及映射后的低維空間中數(shù)據(jù)點之間的相似性相關(guān)的概率分布,然后最小化兩個分布之間的Kullback-Leibler散度。t-SNE被廣泛應(yīng)用于單細胞數(shù)據(jù)分析中的數(shù)據(jù)可視化。
CCA:是一種基于互協(xié)方差矩陣的降維方法。給定兩個或多個數(shù)據(jù)集,該方法查找每個數(shù)據(jù)集的映射,以最大化數(shù)據(jù)集之間的相關(guān)性。在單細胞轉(zhuǎn)錄組的數(shù)據(jù)分析中,CCA通常用于不同來源樣本的整合,如Seurat(圖2)。
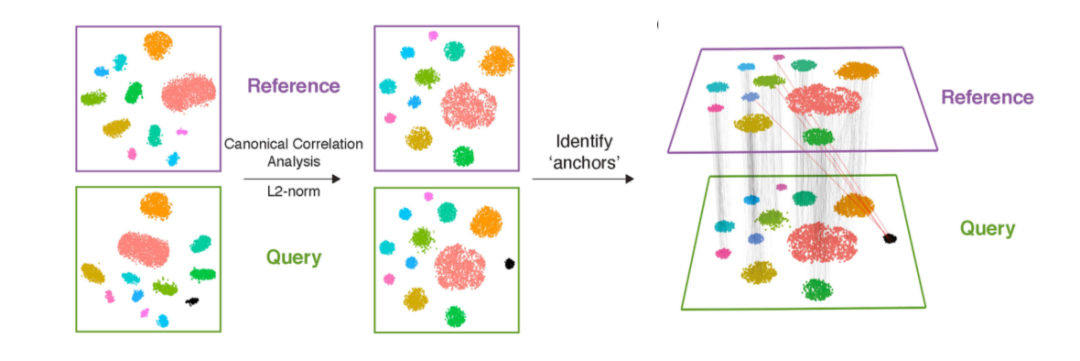
Figure 2. Seurat CCA數(shù)據(jù)整合示意圖
LDA:該方法最初是在自然語言處理中提出的。LDA假設(shè)一個文檔(document)是通過如下方法生成的:首先從具有狄利克雷先驗的話題(topic)的多項分布中對話題進行抽樣,然后對文檔中的單詞(word)進行抽樣,這些單詞的多項式分布是基于每個話題的狄利克雷先驗條件。然后,每個文檔都可以在包含k個話題的低維空間中表示。cellTree使用LDA學習“topics”作為潛在特征來表示細胞,其中“words”是受所選的潛在特征制約的基因表達水平。LDA的生成過程產(chǎn)生了一組可解釋的潛在特征。
相似度及核函數(shù)
在許多聚類方法的計算過程中,不是使用降維的方法,而是通過核函數(shù)或相似度函數(shù)來計算單個細胞之間的配對相似性進行聚類。核函數(shù)策略將從N × M表達矩陣中計算獲得N × N相似矩陣,以期望通過核映射或相似函數(shù)在隱式特征映射空間中減少原始特征空間中的差異(如果使用有效的核函數(shù))。SNN-cliq和Seurat使用SNN作為相似圖。cellTree在用LDA找到的話題直方圖上通過卡方找到細胞間的距離。DTWscore利用時間序列樣本為每個基因找到細胞對之間的動態(tài)時間規(guī)整(DTW)距離,以選擇高度可變的基因,其中DTW距離是基于兩個時間序列在最佳規(guī)整路徑上的比對計算的。基于TCC的聚類使用細胞間的Jensen-Shannon距離作為譜聚類或近鄰傳播聚類的輸入。SIMLR結(jié)合多個核來學習得到細胞相似矩陣,并使用秩約束和圖擴散來解決dropouts問題。
大多數(shù)其他方法使用更標準的相似性函數(shù)或距離函數(shù)。BackSPIN,DendroSplit,ICGS和SINCERA在層次聚類策略中使用Pearson相關(guān)來尋找最佳分割點。GiniClust和RaceID也分別使用相關(guān)性矩陣進行DBSCAN和k-means聚類。參考成分分析(RCA)計算單個細胞和參考細胞之間的表達譜之間的相關(guān)性,作為聚類的新特征,以最小化技術(shù)差異和批次效應(yīng)。SC3使用斯皮爾曼、皮爾森和歐氏距離來計算細胞間的配對相似性或距離以獲得一致性矩陣。
審核編輯:劉清
-
PCR
+關(guān)注
關(guān)注
0文章
120瀏覽量
19664 -
機器學習
+關(guān)注
關(guān)注
66文章
8441瀏覽量
133094 -
RNA
+關(guān)注
關(guān)注
0文章
46瀏覽量
9739 -
UMI
+關(guān)注
關(guān)注
0文章
3瀏覽量
1414
原文標題:單細胞轉(zhuǎn)錄組 | 聚類分析中的機器學習與統(tǒng)計方法綜述(一)
文章出處:【微信號:SBCNECB,微信公眾號:上海生物芯片】歡迎添加關(guān)注!文章轉(zhuǎn)載請注明出處。
發(fā)布評論請先 登錄
相關(guān)推薦
傳統(tǒng)機器學習方法和應(yīng)用指導

zeta在機器學習中的應(yīng)用 zeta的優(yōu)缺點分析
Minitab 在統(tǒng)計分析中的應(yīng)用
什么是機器學習?通過機器學習方法能解決哪些問題?






 工商網(wǎng)監(jiān)
工商網(wǎng)監(jiān)
評論